您好!歡迎訪問德爾塔儀器官方網站




gaoshengkeji@163.com

為加強醫療器械產品注冊工作的監督和指導,進一步提高注冊審查質量,國家藥品監督管理局組織制定了《醫療器械臨床試驗數據遞交要求注冊審查指導原則》《體外診斷試劑臨床試驗數據遞交要求注冊審查指導原則》,現予發布。
特此通告。
附件:1.醫療器械臨床試驗數據遞交要求注冊審查指導原則
2.體外診斷試劑臨床試驗數據遞交要求注冊審查指導原則
國家藥監局
2021年11月25日
附件1
醫療器械臨床試驗數據遞交
要求注冊審查指導原則
一、前言
醫療器械臨床試驗數據是評價醫療器械安全有效性的重要支持性資料之一。規范地收集、整理、分析和遞交醫療器械臨床試驗數據有助于提高臨床試驗實施和管理質量,同時有利于監管機構快速、高效地掌握臨床試驗的開展情況,提高審評效率。
為指導注冊申請人規范遞交臨床試驗數據及相關資料,以便更好地開展臨床評價資料審評相關工作,制定本指導原則。
本指導原則是在現行法規和標準體系以及當前認知水平下制定的,隨著法規和標準的不斷完善,以及科學技術的不斷發展,本指導原則的相關內容也將進行相應的調整。
二、適用范圍
本指導原則適用于以產品注冊為目的開展的醫療器械臨床試驗,包括在境外開展的醫療器械臨床試驗,不適用于按醫療器械管理的體外診斷試劑臨床試驗。與體外診斷試劑配合使用開展臨床試驗的體外診斷設備和軟件,如將該臨床試驗用于該體外診斷設備和軟件的注冊申報,可參考本指導原則提交臨床試驗數據,亦可按照《體外診斷試劑臨床試驗數據遞交要求注冊審查指導原則》提交臨床試驗數據。本指導原則僅涉及臨床試驗數據遞交相關內容,不涉及臨床試驗過程中數據管理相關要求。
三、基本原則
(一)真實原則
所遞交的臨床試驗數據與臨床試驗原始記錄保持一致。
(二)可追溯原則
按照注冊申請人提交的數據、說明性文件和程序代碼,可從原始數據庫重現形成分析數據庫、臨床試驗報告中的統計分析結果,且形成的分析數據庫和統計分析結果與注冊申請人提交的內容一致。
(三)可讀原則
所提交數據庫結構清晰,注釋詳盡,便于審閱。按照本指導原則相關規范要求遞交臨床試驗數據有助于提高可讀性。
四、臨床試驗數據相關資料及其說明
醫療器械臨床試驗數據相關資料通常包括原始數據庫、分析數據庫、說明性文件和程序代碼,以下對各申報資料具體格式和內容提出要求。鼓勵注冊申請人參照臨床數據交換標準協會(Clinical Data Interchange Standards Consortium, CDISC)標準遞交數據。
外文資料提供中文翻譯件時,對于原始和分析數據集,應至少翻譯數據集、變量標簽、觀測值中的描述性文本(如不良事件描述等)。
(一)原始數據庫
遞交的原始數據庫通常包含從病例報告表和外部文件中直接收集的原始數據,缺失的數據在此處不應進行填補。
原始數據庫通常由多個不同的原始數據集組成,單個原始數據集是相同主題下多個變量的集合,這些變量的觀測值共同形成該原始數據集,例如,人口學資料數據集可包括年齡、性別、身體質量指數(Body Mass Index,BMI)等。不同臨床試驗涉及的原始數據庫不完全相同。單個原始數據集應收集相同主題下的變量,不同主題變量建議分別形成原始數據集,例如,膝關節Lysholm評分和IKDC2000評分相關變量建議分別形成兩個原始數據集。
各數據集需包括受試者唯一標識變量,以實現同一受試者不同數據集觀測值的關聯。如涉及不同訪視時間點觀測的數據,應使用訪視時間變量進行標識。例如,術后3個月和6個月心臟超聲相關觀測值,訪視時間標識變量可命名為Visit_3、Visit_6等進行區分。若涉及兩個及兩個以上臨床試驗,數據集中需包括臨床研究標識變量。若臨床試驗采用了隨機分組,原始數據庫中應包含隨機號等變量。
數據集和變量命名應遵循“可讀性”的原則,建議在對其命名時參考數據集或變量的英文或拼音,使得通過名稱可較容易聯想到該命名的實際含義。例如,可參考“Medical History”將病史數據集命名為“MH”,參考“Concomitant Medication”將合并用藥數據集命名為“CM”,將變量“性別”命名為“sex”,變量“受試者姓名縮寫”命名為“sub_abbr”等。
(二)分析數據庫
分析數據庫是為便于統計分析使用原始數據集形成的數據庫,用于產生臨床試驗報告中的統計結果(包括基線、療效和安全性指標統計分析等)。分析數據庫主要包括原始數據庫中的變量數據和按照臨床試驗方案和統計分析計劃(如有)中事先確定的方法(如缺失值填補、量表子項評分加和等)從原始數據庫變量數據衍生的數據。
分析數據庫通常由多個不同的數據集組成,這些數據集一般與臨床試驗報告中的統計結果相對應。例如,臨床試驗報告中美國國立衛生研究院卒中量表(NIHSS)評分統計結果可對應專門的分析數據集,該分析數據集是為了生成NIHSS評分統計結果而專門創建的,包含生成該統計結果的全部變量數據,其他不相關變量數據不納入到該數據集中。為了便于統計分析復驗,分析數據集中的變量應具有可溯性,變量結構應清晰,不需繁瑣的數據前處理即可開展統計分析。
分析數據集可基于其產生的相應統計結果進行命名,例如,生成不良事件比較結果的數據集可命名為“ADAE”(不良事件分析數據集)。建議在分析數據集名稱中加入“AD”(analysis data)前綴,以標識該數據集為分析數據集。
分析數據集變量命名同原始數據庫要求。注意明確不同分析集(如全分析集FAS、符合方案集PPS和安全分析集SS等)標識變量,以及形成數據庫過程中產生的系統變量(如有),如序號、時間等。
(三)程序代碼
需遞交的代碼主要包括:用于原始數據庫生成分析數據庫的代碼、分析數據庫生成統計分析結果的代碼等,用于調整格式或生成表格的相關代碼可不遞交。遞交的代碼應符合通常的編程格式和編程規范,結構清晰明了,易于閱讀。程序代碼中應包括充分的注釋,以描述不同程序代碼的目的及其他需解釋的內容,幫助審閱者更好地理解代碼邏輯。如果遞交的程序代碼引用了宏程序,需提供相應的宏程序代碼,并說明運行該程序的軟件版本、系統環境。
(四)說明性文件
1.數據說明文件
數據說明文件用于描述原始數據庫和分析數據庫的內容和結構,有助于審閱者快速了解數據庫中各數據集、變量及其相互間的結構關系,準確理解遞交的數據內容。建議采用Excel文件,以表格的形式分別列明原始數據庫和分析數據庫中所含的數據集、變量、變量類型(如字符型、數值型)、標簽、賦值及其對應關系,具體可參考附錄1《數據集、變量關系列表舉例》。為了便于審閱,數據集和變量應具有相應的中文標簽,標簽長度不宜過長。若使用了外部詞典(如MedDRA),應明確使用的外部詞典名稱和版本號。
分析數據庫的說明文件需列明衍生變量的生成規則,明確涉及到的變量和計算方法。例如,對缺失值的填補,應明確填補方法,提供相應的程序代碼。建議以表格的形式列明生成各分析數據集所用到的程序代碼文件和原始數據集名稱。
2.程序代碼使用說明文件
程序代碼使用說明文件用來解釋說明程序代碼文件使用方法、系統及軟件環境,包括使用代碼文件時是否需修改以及如何修改程序代碼。同時,以表格形式逐一列明生成各統計結果圖表所使用到的程序代碼文件和數據集文件名稱。
注冊申請人應說明原始數據集和分析數據集所用編碼(如UTF-8、EUC-CN等),以避免所遞交的數據集出現亂碼的情形。
3.注釋病例報告表
相對于空白CRF,注釋CRF增加了注釋內容,反映了數據庫中變量與CRF信息收集的對應關系。例如,在性別空白處注釋變量名稱sex。利用注釋CRF,審閱者可直觀地查閱各變量在CRF中的位置。CRF中可能收集了一些與臨床試驗結果分析無關的冗余數據,這些數據可不包含在遞交的數據庫中,但應在注釋CRF上明確標注為“不遞交”,并闡明理由。
4.其他說明性文件
除以上說明性文件外,鼓勵注冊申請人提交其他有利于審閱者快速了解臨床試驗數據庫內容和結構的說明性文件(如概覽性文件、其他特殊情況說明文件等)。
五、遞交形式
原始數據庫、分析數據庫、說明性文件和程序代碼分別放置于四個文件夾中。
原始數據庫和分析數據庫建議采用XPT數據傳輸格式遞交,建議全部原始數據集形成一個XPT文件,全部分析數據集形成一個XPT文件。建議采用XPT第5版本(簡稱XPT V5)或以上版本作為數據遞交格式。
數據說明文件可采用PDF、Word、Excel等文件格式,其中變量詞典建議采用Excel文件,注釋病例報告表建議采用PDF文件。
程序代碼建議采用TXT文件格式。
六、起草單位
國家藥品監督管理局醫療器械技術審評中心。
附錄1
數據集、變量關系列表舉例
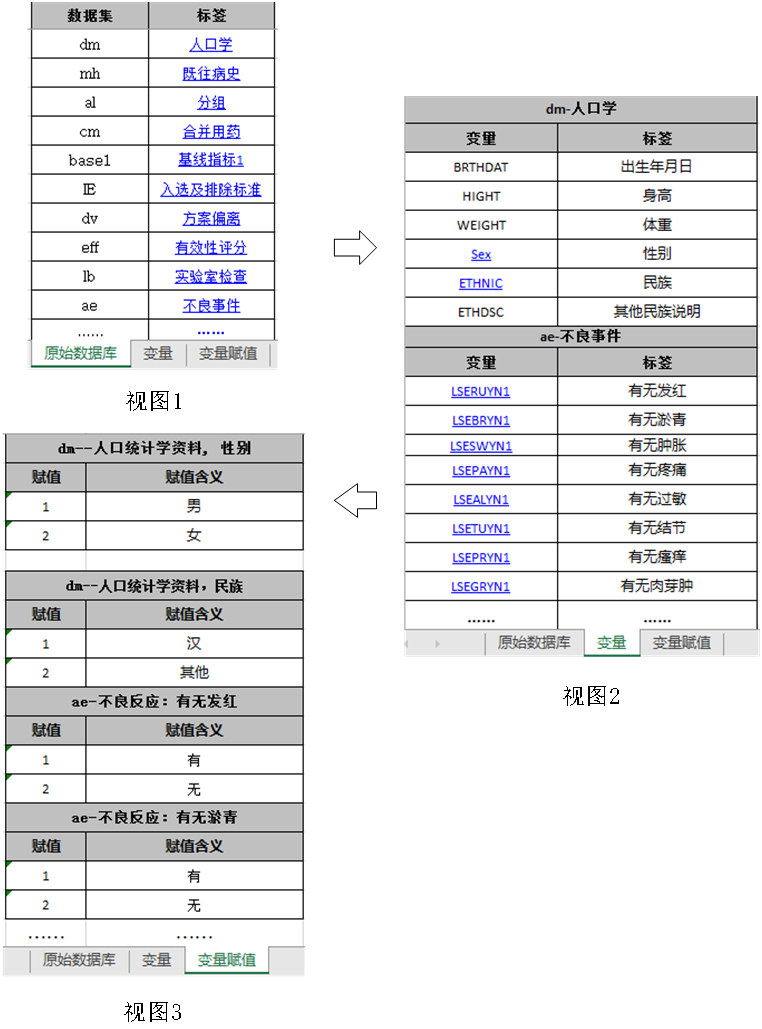
注:有下劃線文字帶有超鏈接,點擊可直接跳轉到鏈接位置。例如,點擊視圖1中“人口學”,可直接跳轉至視圖2中“人口學”數據集所含變量視圖,點擊視圖2中“sex”,可直接跳轉至視圖3中查看性別具體賦值情況。與變量無關的內容不放入表中。
附件2
體外診斷試劑臨床試驗數據遞交要求
注冊審查指導原則
一、前言
體外診斷試劑臨床試驗數據是評價產品安全有效性的重要支持性資料之一。規范地收集、整理、分析和遞交臨床試驗數據有助于提高臨床試驗實施和管理質量,同時有利于監管機構快速、高效地掌握臨床試驗的開展情況,提高審評效率。
為指導注冊申請人規范遞交體外診斷試劑臨床試驗數據及相關資料,以便更好地開展臨床試驗資料審評相關工作,制定本指導原則。
本指導原則是在現行法規和標準體系以及當前認知水平下制定的,隨著法規和標準的不斷完善,以及科學技術的不斷發展,本指導原則的相關內容也將進行相應的調整。
二、適用范圍
本指導原則適用于以產品注冊為目的開展的體外診斷試劑臨床試驗,包括在境外開展的體外診斷試劑臨床試驗。本指導原則僅涉及臨床試驗數據遞交相關內容,不涉及臨床試驗過程中數據管理相關要求。
三、基本原則
(一)真實原則
所遞交的臨床試驗數據應與臨床試驗中所有的原始記錄保持一致。
(二)可追溯原則
應能夠根據注冊申請人所提交的數據、說明性文件和程序代碼(如有),可從原始數據庫重現形成分析數據庫、臨床試驗報告中的統計結果,且形成的分析數據庫和統計分析結果與注冊申請人提交的內容一致。
注冊申請人提交的臨床試驗數據庫,應可溯源至臨床試驗中的原始試驗記錄、病例報告表,臨床背景信息應可追溯至臨床試驗機構的病例相關信息管理系統或原始記錄。原始試驗記錄可能包括樣本的篩選入選表、樣本編盲表、檢測記錄表等,如申報產品需要配套儀器進行檢測,則還應包括配套儀器上的電子記錄。以上原始試驗記錄不要求遞交,應妥善保管備查。
(三)數據全面且可讀原則
所提交的臨床試驗數據根據申報產品的特點以及臨床試驗設計的不同,應保證數據全面、可讀,且易于統計。數據庫結構清晰、注釋詳盡,便于審閱。
四、臨床試驗數據庫相關內容要求
一般來說,臨床試驗數據庫相關內容應包括受試者相關信息以及臨床試驗樣本的檢測信息。不同的臨床試驗設計類型所應包含的數據信息亦不相同。根據產品特點和產品性能評價需要,體外診斷試劑臨床試驗包括不同的臨床試驗設計類型,根據在臨床試驗過程中試驗體外診斷試劑檢測結果對受試者的影響,一般分為觀察性研究和干預性研究。其中觀察性研究根據臨床試驗中所檢測的不同時間點又可分為橫斷面研究和縱向研究。橫斷面研究是體外診斷試劑臨床試驗最常見的設計類型,其臨床試驗數據庫內容的要求亦是體外診斷試劑臨床試驗數據庫內容的通用要求。對于縱向研究以及干預性研究應在通用要求的基礎上再補充相應的數據。
(一) 來自于橫斷面研究的數據
臨床試驗中的入組人群、樣本類型等均應與申報產品所聲稱的預期用途保持一致。因此數據信息中應包括受試者相關信息,包括臨床診斷背景信息、樣本類型、人口學信息(性別、年齡等)等。臨床診斷背景信息包括臨床診斷結果、相關的癥狀體征、以及診療信息(如需要)等。如申報產品的統計分析中需要進行亞組統計,則應包括與亞組劃分相關的信息,如疾病的不同分期、不同進程等信息。
臨床試驗中關于臨床樣本的檢測信息主要包括:試驗體外診斷試劑的檢測結果、對比方法檢測結果等。對于根據所確定的陽性判斷值來判定檢測結果的產品,數據信息還應包括試驗體外診斷試劑和對比方法的詳細檢測數值(如Ct值、S/CO值等)。核酸檢測類產品如涉及不同檢測通道的,各通道的檢測數值均應提供,包括內標的檢測數值。
臨床試驗數據應真實且可追溯,數據集中應有唯一可溯源的去標識化樣本編號,該樣本編號應能夠溯源至該病例的所有背景信息,如病例編號、診療信息等。
如臨床試驗中涉及復測等情況,相應的數據集中應有初測及復測的數據,并備注復測原因。如有其他需要說明的信息,可增加一列“備注”,將信息增加至“備注”中。
(二) 來自于縱向研究的數據
除橫斷面研究外,有些產品需要進行縱向研究,對于縱向研究的數據,數據集應包括每個病例的每個時間點的具體數據,應盡量將同一病例的每一時間點的數據匯總,采樣時間點和檢測結果應對應列出。對于此種臨床試驗設計,會涉及多個樣本來自于同一個受試者的情形,則應同時提供去標識化的受試者編號和樣本編號。
(三)來自于干預性研究的數據
對于干預性研究的數據,數據集中除以上基本信息外,還包括病例的臨床試驗分組、具體的診療信息、病例的臨床評價終點等信息。
五、遞交形式
體外診斷試劑臨床試驗數據相關的申報資料通常包括原始數據庫、分析數據庫、說明性文件和程序代碼(如有),以下對各申報資料具體格式和內容提出要求。鼓勵注冊申請人參照臨床數據交換標準協會(Clinical Data Interchange Standards Consortium,CDISC)標準遞交數據。
建議注冊申請人綜合考慮臨床試驗電子數據采集系統/數據管理系統(Electronic Data Capture System,EDC)在數據采集及管理方面的優勢,逐步推進EDC系統的使用,尤其對于數據量較大、產品風險較高的臨床試驗。
外文資料提供中文翻譯件時,需注意對于原始和分析數據集,至少應翻譯數據集、變量標簽、觀測值中的描述性文本。
(一)原始數據庫
原始數據庫通常包含從病例報告表和外部文件中直接收集的原始數據,應包括臨床試驗按照方案的要求入組的所有病例及樣本信息,原始數據庫中的缺失數據不應進行填補。按照臨床試驗方案的剔除標準進行剔除的病例亦應包括在內,同時應備注剔除原因。
對于數據量較大的臨床試驗,原始數據庫通常由多個不同的原始數據集組成。單個原始數據集是相同主題下多個變量的集合,這些變量的觀測值共同形成該原始數據集。例如,人口學資料數據集可包括去標識化的受試者編號、年齡、性別、臨床診斷等。不同臨床試驗涉及的原始數據庫不完全相同。單個原始數據集應收集相同主題下的變量。各數據集需包括受試者唯一標識變量,以實現同一受試者不同數據集數值之間的關聯。若臨床試驗采用了隨機分組,原始數據庫中應包含隨機號等變量。
數據集和變量命名應遵循“可讀性”的原則,建議在對其命名時參考數據集或變量的英文或拼音,通過名稱可較容易聯想到該命名的實際含義。數據集通常以兩個字母組成的代碼命名,如受試者人口學數據集(dm)、樣本檢測數據集(lb)、樣本采集與處理數據集(cl)、不良事件(ae)等。將變量“性別”命名為“Sex”、“年齡”命名為“Age”、“受試者編號”命名為“SUBJID”、“樣本編號”命名為“SAMID”、“臨床診斷”命名為“DIAG”等。
(二)分析數據庫
分析數據庫是為便于統計分析使用原始數據集形成的數據庫,用于產生臨床試驗報告中的統計結果。分析數據庫主要包括原始數據庫中的變量數據和按照臨床試驗方案或統計分析計劃(如有)中事先確定的方法使用原始數據庫變量數據衍生的數據。
分析數據庫通常由多個不同的數據集組成,其中的數據集形成應與臨床試驗報告中的統計結果相對應。如需進行亞組分析時,可針對亞組分析構建不同的分析數據集。對于定量檢測類試劑,需要對檢測數據在線性范圍內的樣本納入定量相關分析,則應針對線性范圍內的所有樣本生成分析數據集。為了便于統計分析復驗,分析數據集中的變量應具有可溯性,變量結構應清晰,不需繁瑣的數據前處理即可開展統計分析。
分析數據集可基于其產生的相應統計結果進行命名,建議在分析數據集名稱中加入“AD”(analysis data)前綴,以標識該數據集為分析數據集。
(三)說明性文件
1.數據說明文件
數據說明文件用于描述原始數據庫和分析數據庫的內容和結構,是助于審閱者快速了解數據庫中各數據集、變量及其相互間的結構關系,準確理解遞交的數據內容。建議采用Excel文件,以表格的形式列明原始數據庫和分析數據庫中所含的數據集、變量、變量類型(如字符型、數值型)、標簽、賦值及其對應關系。為了便于審閱,數據集和變量應具有相應的中文標簽,標簽長度不宜過長。若使用了外部詞典(如MedDRA),應明確使用的外部詞典名稱和版本號。
分析數據庫的說明文件需列明衍生變量的生成規則,明確涉及到的變量和計算方法。如注冊申請人使用程序代碼生成分析數據庫,建議以表格的形式列明生成各分析數據集所用到的程序代碼文件和原始數據集名稱。
2.統計分析說明文件
注冊申請人應將從分析數據庫到最終生成臨床試驗報告中的統計結果的過程或計算方法詳細列明。
如統計分析的過程直接在Excel中實現的,應在說明性文件中將所用的函數公式列明。如使用統計工具進行統計分析但未使用程序代碼的,可以文字加圖示的形式詳細闡明統計分析步驟,應注明所用統計工具的版本號。
統計分析中使用程序代碼的,應在統計分析說明文件中解釋說明程序代碼文件的使用方法、系統及軟件環境,包括使用代碼文件時是否需修改以及如何修改程序代碼。同時,以表格形式逐一列明生成各統計結果圖表所使用到的程序代碼文件和數據集文件名稱。注冊申請人應說明原始數據集和分析數據集所用編碼(如UTF-8、EUC-CN等),以避免所遞交的數據集出現亂碼的情形。同時應參照以下第(四)條要求提供相應的程序代碼。
3.注釋病例報告表(如有)
相對于空白CRF,注釋CRF增加了注釋內容,反映了數據庫中變量與CRF表信息收集的對應關系。例如,在性別空白處注釋變量名稱Sex。利用注釋CRF,審閱者可直觀地查閱各變量在CRF表中的位置,CRF中可能收集了一些與臨床試驗結果分析無關的冗余數據,這些數據可不包含在遞交的數據庫中,但應在注釋CRF上明確標注為“不遞交”,并闡明理由。
4.其他說明性文件
除以上說明性文件外,鼓勵注冊申請人提交其他有利于審評人員快速了解臨床試驗數據庫內容和結構的說明性文件(如概覽性文件、其他特殊情況說明文件等)。
(四)程序代碼(如有)
如數據庫的管理或統計分析中使用程序代碼的,應提供程序代碼,需遞交的代碼主要包括:用于原始數據庫生成分析數據庫的代碼、分析數據庫生成統計結果的代碼等,用于調整格式或生成表格的相關代碼不需遞交。遞交的代碼應符合通常的編程格式和編程規范,結構清晰明了,易于閱讀。代碼中以中文注釋的形式描述不同程序模塊的目的以及其他需解釋的內容。如提交的代碼中引用了宏程序,則需要提供相應的宏程序代碼,并說明可以運行該程序的軟件版本和系統環境。
(五)形式要求
原始數據庫、分析數據庫、說明性文件和程序代碼(如有)分別放置于四個文件夾中。
臨床試驗數據庫可以Excel形式進行遞交,如使用EDC系統進行數據采集及管理的,原始數據庫和分析數據庫建議采用XPT1數據傳輸格式進行遞交,建議全部原始數據集形成一個XPT文件,全部分析數據集形成一個XPT文件。建議采用XPT第5版本(簡稱XPT V5)或以上版本作為數據遞交格式。
數據說明文件及統計分析說明文件采用PDF、Word、Excel文件,其中變量詞典建議采用Excel文件,注釋病例報告表建議采用PDF文件。
程序代碼建議采用TXT文件。
六、起草單位
國家藥品監督管理局醫療器械技術審評中心
【來源】國家藥品監督管理局


二維碼(微信公眾號)

二維碼(新浪微博)
 關閉返回
關閉返回